6.5 KiB
- Biology Meets Programming: Bioinformatics for Beginners
Biology Meets Programming: Bioinformatics for Beginners
Week 1
DNA replication
Origin of replication (ori)
Locating an ori is key for gene therapy (e.g. viral vectors), to introduce a theraupetic gene.
Computational approaches to find ori in Vibrio Cholerae
Exercise: find Pattern
We'll look for the DnaA box sequence, using a sliding window, in that case we will use the function PatternCount to find out how many times does a sequence appear in the genome.
For the second part, we're going to calculate the frequency map of the sequences of length k, for that purpose we'll use FrequentWords
Exercise: Find the reverse complement of a sequence
We're going to generate the reverse complement of a sequence, which is the complement of a sequence, read in the same direction (5' -> 3'). In this case, we're going to use ReverseComplement After using our function on the Vibrio Cholerae's genome, we realize that some of the frequent k-mers are reverse complements of other frequent ones.
Exercise: Find a subsequence within a sequence
We're going to find the ocurrences of a subsquence inside a sequence, and save the index of the first letter in the sequence. This time, we'll use PatternMatching After using our function on the Vibrio Cholerae's genome, we find out that the 9-mers with the highest frequency appear in cluster. This is strong statistical evidence that our subsequences are DnaA boxes.
Computational approaches to find ori in any bacteria
Now that we're pretty confident about the DnaA boxes sequences that we found, we are going to check if they are a common pattern in the rest of bacterias. We're going to find the ocurrences of the sequences in Thermotoga petrophila using PatternCount
After the execution, we observe that there are no ocurrences of the sequences found in Vibrio Cholerae. We can conclude that different bacterias have different DnaA boxes.
We have to try another computational approach then, find clusters of k-mers repeated in a small interval.
Week 2
DNA replication (II)
Replication process
The DNA polymerases start replicating while the parent strands are unraveling. On the lagging strand, the DNA polymerase waits until the replication fork opens around 2000 nucleotides, and because of that it forms Okazaki fragments. We need 1 primer for the leading strand and 1 primer per Okazaki fragment for the lagging strand. While the Okazaki fragments are being synthetized, a DNA ligase starts joining the fragments together.
Computational approach to find ori using deamination
As the lagging strand is always waiting for the helicase to go forward, the lagging strand is mostly in single-stranded configuration, which is more prone to mutations. One frequent form of mutation is deamination, a process that causes cytosine to convert into thymine. This means that cytosine is more frequent in half of the genome.
Exercise: count the ocurrences of cytosine
We're going to count the ocurrences of the bases in a genome and include them in a symbol array, for that purpose we'll use SymbolArray After executing the program, we realize that the algorithm is too inefficient.
Exercise: find a better algorithm for the previous exercise
This time, we are going to evaluate an element i+1, using the element i. We'll use FasterSymbolArray to achieve this After executing the program we see that it's a viable algorithm, with a complexity of O(n) instead of the previous O(n²). In Escherichia Coli we plotted the result of our program:
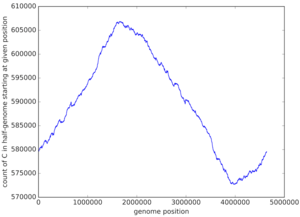
From that graph, we conclude that ori is located around position 4000000, because that's where the Cytosine concentration is the lowest, which indicates that the region stays single-stranded for the longest time.
The Skew Diagram
Usually scientists measure the difference between G - C, which is higher on the lagging strand and lower on the leading strand.
Exercise: Synthetize a Skew Array
We're going to make a Skew Diagram, for that we'll first need a Skew Array. For that purpose we wrote SkewArray We can see the utility of a Skew Diagram looking at the one from Escherichia Coli:
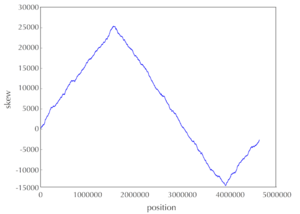
Ori should be located where the skew is at its minimum value.
Exercise: Efficient algorithm for locating ori
Now that we know more about ori's skew value, we're going to construct a better algorithm to find it. We'll do that in MinimumSkew
Finding DnaA boxes
When we look for DnaA boxes in the minimal skew region, we can't find highly repeated 9-mers in Escherichia Coli. But we find approximate sequences that are similar to our 9-mers and only differ in 1 nucleotide.
Exercise: Calculate Hamming distance
The Hamming distance is the number of mismatches between 2 strings, we'll solve this problem in HammingDistance
Exercise: Find approximate patterns
Now that we have our Hamming distance, we have to find the approximate sequences. We'll do this in ApproximatePatternMatching.py
Exercise: Count the approximate patterns
The final part is counting the approximate sequences, for that we'll use ApproximatePatternCount.py
After trying out our ApproximatePatternCount in the hypothesized ori region, we find a frequent k-mer with its reverse complement in Escherichia Coli. We've finally found a computational method to find ori that seems correct.
Week 3
The circadian clock
Variation in gene expression permits the cell to keep track of time.
Computational approaches to find regulatory motifs
Vocabulary
- k-mer: subsquences of length k in a biological sequence
- Frequency map: sequence –> frequency of the sequence